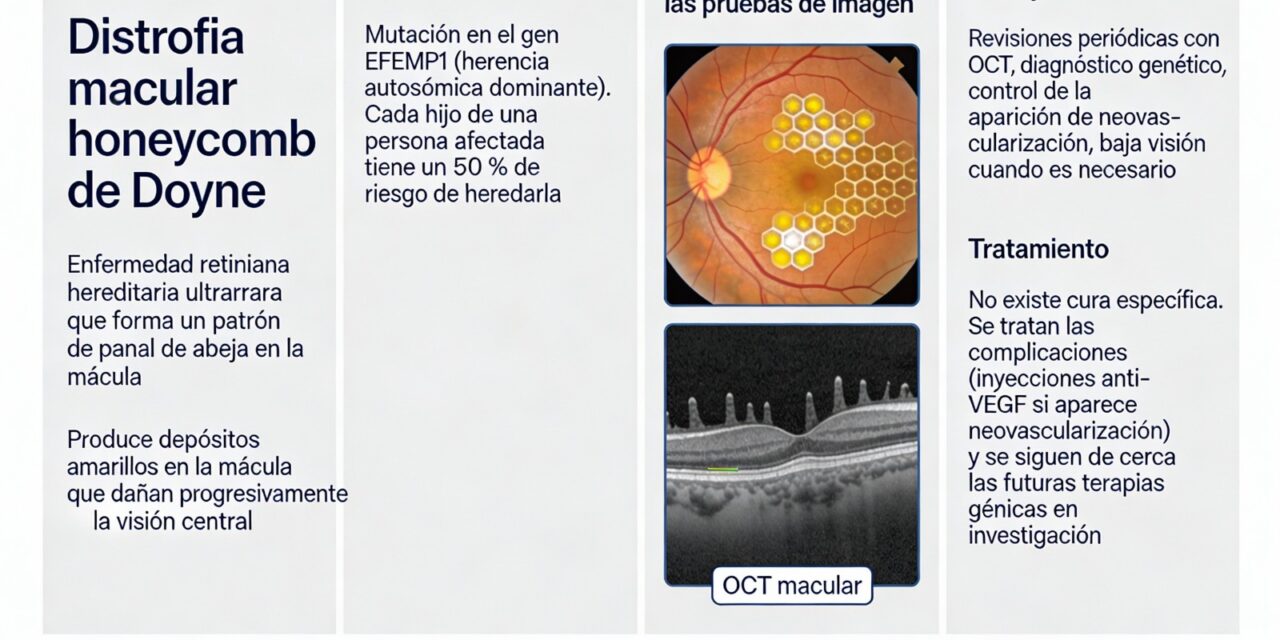
Panal de abeja. Así se traduce honeycomb, una enfermedad de la retina considerada ultrarrara que padecen muy pocas personas en el mundo, muchas de ellas sin saberlo. Su definición completa es ‘distrofia retiniana honeycomb de Doyne’ y su nombre obedece a que los depósitos que la caracterizan forman un patrón similar a los hexágonos de un panal de miel en la parte trasera del ojo.
Los oftalmólogos diagnostican esta minoritaria enfermedad cuando observan en el fondo del ojo pequeños depósitos amarillo-blanquecinos que se distribuyen en la mácula—la zona responsable de la visión central—formando ese patrón característico. Estos depósitos destruyen lentamente la vista, robando poco a poco la capacidad de distinguir bien la claridad.
Lo insidioso es que es tan desconocida que incluso muchos oftalmólogos la confunden con otras enfermedades más comunes. Por eso son cada vez más determinantes las pruebas genéticas para confirmar diagnósticos. Es fundamental también para que los primeros ensayos de terapias génicas que un grupo de investigadores de Londres confía en tener listos en 2-5 años tengan bien definidas buenas cohortes de pacientes. Y dimensionado correctamente el universo de afectados por la enfermedad.
Minoritaria hasta la invisibilidad
La honeycomb es causada por una mutación en un único gen llamado EFEMP1. Solo se necesita heredar una copia defectuosa de ese gen para desarrollar la enfermedad, lo que significa que, si un progenitor la tiene, los descendientes tienen un 50% de probabilidad de heredarla.
Este hecho contrasta con que los registros científicos más completos documentan apenas 44 pacientes confirmados genéticamente en el mundo. Supone que no hay una base fiable para esta enfermedad y que no existen datos o estudios sistematizados; como por otro lado ocurre con otras muchas enfermenades raras. Se estima que hay cientos a pocos miles de afectados globalmente, convirtiéndola en una de las distrofias retinianas más raras conocidas.
Comparativamente, la enfermedad de Stargardt—otra distrofia macular hereditaria—afecta a una de cada 8.000-10.000 personas. La enfermedad de Best, que también causa depósitos en la mácula, afecta a una de cada 10.000. Incluso el síndrome de Kearns-Sayre, extremadamente raro, tiene más pacientes documentados que honeycomb. Eso ilustra el nivel de rareza de esta condición.
El problema del diagnóstico confuso
Uno de los motivos es que honeycomb es casi imposible de distinguir a primera vista de la degeneración macular relacionada con la edad (DMAE), que es mucho más común. Ambas muestran depósitos bajo la retina, pero con causas completamente diferentes. DMAE es adquirida por envejecimiento e inflamación; honeycomb es genética y heredada desde el nacimiento, manifestándose alrededor de los 40-50 años.
Además de DMAE, los oftalmólogos deben descartar la enfermedad de Stargardt, que causa manchitas características distribuidas por toda la mácula, pero típicamente comienza en la adolescencia, no en la mediana edad. También está la enfermedad de Best, que produce depósitos más grandes con forma de «yema de huevo» amarillenta, claramente distinta del patrón hexagonal honeycomb. El síndrome de Alport puede causar una maculopatía parecida a panal, pero siempre acompaña con insuficiencia renal progresiva y pérdida auditiva, diferenciándola inmediatamente.
Los pacientes con honeycomb experimentan visión borrosa central progresiva, dificultad para distinguir colores, líneas que se ven onduladas, y problemas adaptándose de la luz a la oscuridad por falta de sensibilidad al contraste. Mientras los depósitos avanzan durante años, aproximadamente un tercio de los pacientes nunca desarrollan síntomas reales, por lo que pueden tener la enfermedad sin saberlo.
Ningún ensayo clínico ni tratamiento
Tal y como suele ocurrir con la mayoría de las enfermedades raras, no existe actualmente ni un solo ensayo clínico específico para honeycomb en reclutamiento activo. Los pacientes con complicaciones de neovascularización coroidal—que ocurren en una de cada cuatro personas con honeycomb—reciben las mismas inyecciones anti-VEGF (antiangiogénicos) que se usan para la DMAE o el edema macular diabético. No hay tratamiento aprobado que detenga la progresión de los depósitos.
Lo único disponible es vigilancia oftalmológica regular, asesoramiento genético para familias, y esperar que las complicaciones no aparezcan.
Terapia génica en desarrollo
Pero las nuevas técnicas de desarrollo de terapias génicas están trayendo movimiento y primeras esperanzas, igual que a otras distrofias hereditarias de la retina hasta ahora sin perspectiva alguna. La Universidad de Londres cuenta con un programa financiado por la Fundación Macular para desarrollar un tratamiento revolucionario basado en oligonucleótidos antisentido, una tecnología que «apaga» el gen defectuoso permitiendo que el gen sano haga su trabajo. Como honeycomb es autosómica dominante, este enfoque es teóricamente perfecto: silencia la copia rota, activa la copia buena.
Este proyecto está actualmente en fase preclínica, pero según las fuentes consultadas sería el único esfuerzo específico documentado para honeycomb en el mundo. Los expertos esperan que los primeros ensayos clínicos en humanos puedan iniciarse dentro de 2-5 años.
Mientras tanto, se estudia si investigaciones en terapia génica para otras distrofias retinianas podrían ser transferibles. Luxturna, el único tratamiento genético aprobado por la EMA (Agencia Europea del Medicamento) para distrofias retinianas (2018), demostró que entregar genes a la retina es seguro. BEST1 Gene Therapy, para la enfermedad de Best—también autosómica dominante como honeycomb—acaba de recibir aprobación de la FDA norteamericana (Food and Drug Administration) para ensayos Fase 1/2 en 2025. Las terapias con células madre y los inhibidores del complemento están en desarrollo.
Diagnóstico como base
Para los afectados por honeycomb, el diagnóstico genético es crucial. No solo confirma la condición, abriendo acceso a vigilancia oftalmológica regular y consejo genético, sino que también permite a los pacientes participar en futuras investigaciones clínicas cuando estén disponibles. En una enfermedad tan rara que permanece invisible incluso para la comunidad médica, simplemente saber que se padece honeycomb —en lugar de «probablemente” DMAE—es el primer paso hacia la esperanza.
Artículo de elaboración propia, con ayuda de IA para análisis, y síntesis de documentos, así como para la redacción del artículo.
Fuentes: National Organization for Rare Disorders, Universidad de Londres – Proyecto Terapia Génica, Guimarães et al. 2024 – Estudio Historia Natural, EyeWiki – Doyne Honeycomb, Macular Society

{kind=link}