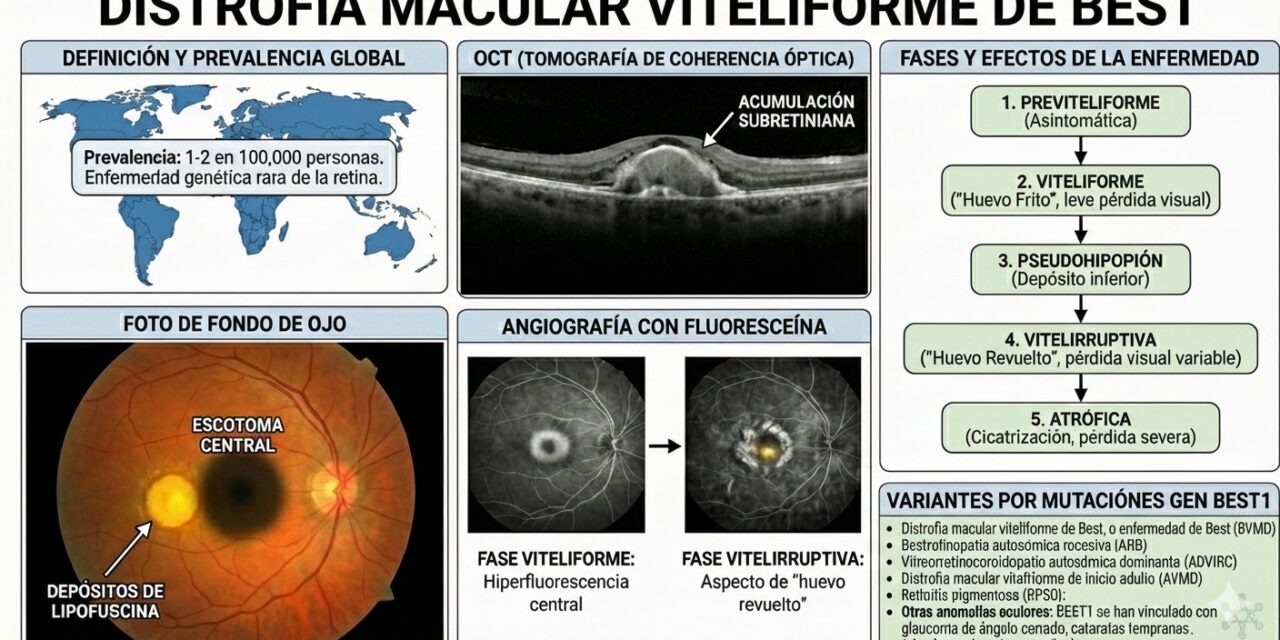
En Europa hay estudios de historia natural y caracterización génica que sientan las bases para futuros tratamientos
La enfermedad de Best, o distrofia macular viteliforme de Best (BVMD), atraviesa un HITO histórico. Por primera vez se conoce que una terapia génica específica contra esta enfermedad hereditaria de la retina entra en fase de evaluación clínica con pacientes. Se trata de OPGx-BEST1, desarrollada por la empresa estadounidense Opus Genetics, que ha anunciado el reclutamiento de sus primeros participantes en el ensayo clínico BIRD-1 (Safety and Tolerability of Subretinally Injected OPGx-BEST1), un estudio que durará hasta 2030 según su registro en ClinicalTrials.gov.
Un hito en la terapia génica ocular
El anuncio fue realizado el 8 de enero de 2026 tras lograr un hito crucial: el primer paciente fue tratado en noviembre de 2025. Desde entonces, cuatro personas han recibido la inyección subretiniana de OPGx-BEST1, con el objetivo inmediato de establecer seguridad y tolerabilidad del tratamiento. En diciembre de 2025, un comité independiente de monitoreo de datos evaluó los resultados iniciales del primero tras un mes de seguimiento y emitió una recomendación positiva para continuar dosificando a los participantes restantes sin modificaciones del protocolo.
OPGx-BEST1 es una terapia basada en un vector adeno-asociado (AAV) que entrega una copia funcional del gen BEST1 directamente a las células del epitelio pigmentario retiniano (EPR), donde reside el gen defectuoso. El ensayo, de naturaleza abierta y adaptativa, evalúa dos cohortes de dosis escalonadas, comenzando con 1,5 mil millones de partículas virales por ojo (Cohorte 1) y una posible escalada a 4,5 mil millones (Cohorte 2). El tratamiento es de una sola inyección y el seguimiento se extiende durante 5 años.
Primeros resultados esperados en 2026
Opus Genetics ha anunciado que presentará datos iniciales de seguridad y actividad biológica en el primer trimestre de 2026 en la Macula Society, y resultados más completos de toda la Cohorte 1 a los 3 meses están previstos para mediados de 2026. La compañía prevé además solicitar designaciones regulatorias especiales en 2026, como Orphan Drug (medicamento huérfano), que aceleraría el desarrollo del tratamiento.
Por el momento, el ensayo se limita a centros estadounidenses, principalmente en Texas, sin planes de expansión anunciados a Europa. Esto contrasta con los esfuerzos europeos, que se centran en entender mejor la enfermedad mediante estudios de historia natural y caracterización molecular.
Investigación fundamental en Europa
En la UE, la iniciativa científica más relevante es el estudio de historia natural NCT05809635, liderado desde la Universidad de Tübingen (Alemania) con colaboración de centros en París (Francia) y en EEUU. Este estudio observacional, activo desde 2023, recluta pacientes con mutaciones confirmadas en BEST1 para caracterizar la progresión natural de la enfermedad mediante evaluaciones anuales de visión funcional (agudeza visual ETDRS, microperimetría MAIA), función electrorretinográfica (ERG/EOG) y estructura retiniana (OCT, autofluorescencia). Los datos son cruciales para definir endpoints sensibles en futuros ensayos terapéuticos europeos y para permitir que el ojo contralateral sin tratar actúe como control interno en estudios unilaterales como BIRD-1.
Recientemente, investigadores franceses del CHU de Lille publicaron un análisis exhaustivo de 450 pacientes franceses con variantes BEST1, identificando 150 variantes distintas, 40 de ellas nuevas. El trabajo, publicado en Investigative Ophthalmology & Visual Science (septiembre 2025), destaca variantes recurrentes específicas en la población francesa, como p.(Ile230del) (3,8% de pacientes) y p.(Arg218His) (8,3%), reforzando la importancia de caracteres específicamente poblacionales en la patogénesis de bestrofinopatías.
Además, el profesor Dr. Bernhard H.F. Weber, del Instituto de Genética Humana de la Universidad de Ratisbona (Alemania), dirige desde al menos dos décadas una investigación centrada en las mutaciones del gen BEST1 y su trabajo se centra en enfoques preclínicos innovadores.
Su grupo ha desarrollado modelos in vitro a partir de células madre pluripotentes inducidas (iPSC) de pacientes para estudiar el epitelio pigmentario retinal (RPE), el sitio principal de patología. Utilizan estas plataformas para dos proyectos de investigación terapéuticos.
El primero se refiere al screening de alto rendimiento (HTS) de compuestos que potencien o corrijan la proteína mutada BEST1, y el segundo a edición génica para eliminar la copia mutada preservando la normal. El último proyecto del Webber registrado en Orphanet se denomina Eliminación específica de alelos de transcritos patogénicos de bestrophin-1 mediante edición genómica dirigida por CRISPR/Cas9.
Edición génica de precisión en España
En España, investigadores del grupo Miranza han presentado resultados prometedores en el campo de la edición génica. La doctora Esther Pomares, responsable de investigación básica de Miranza, lideró un equipo que utilizó técnicas CRISPR y TALEN para corregir con precisión mutaciones causantes de distrofias hereditarias de retina, incluyendo enfermedad de Best, en células madre pluripotentes inducidas (iPSCs) de pacientes. Según explicó la Dra. Pomares, han logrado «revertir» las variantes patogénicas de forma precisa sin alterar otras zonas del genoma, demostrando la validez de ambas tecnologías como herramientas para diseñar futuras terapias personalizadas. Estos avances fueron presentados durante EURETINA 2025 y publicados en la revista Molecular Therapy Nucleic Acids.
Investigación más allá de las pruebas clínicas
En Estados Unidos, el Prof. Alan D. Marmorstein, Ph.D., de la Mayo Clinic en Rochester (Minnesota), ha desarrollado durante años modelos de enfermedad de Best usando células madre derivadas de pacientes. Su equipo ha creado células epiteliales pigmentarias (iPSC-RPE) y un andamio biodegradable para evaluar la posibilidad de trasplante celular. Aunque el protocolo concluyó en 2023, sus trabajos proporcionan evidencia preclínica de que el reemplazo de células RPE defectuosas podría preservar o restaurar la visión, fundamentando estrategias complementarias a la genoterapia.
De forma paralela, el National Institutes of Health (NIH) en Bethesda mantiene activo desde 2007 un ambicioso programa de recopilación de muestras y datos clínicos de pacientes con enfermedades hereditarias raras de retina, incluyendo Best. Este biobanc continuo, actualmente identificado como NCT06491615 en el registro clínico, amplía año a año el archivo de datos genéticos, imágenes y perfiles clínicos que alimentan la investigación global y permiten identificar cohortes de pacientes elegibles para ensayos terapéuticos futuros.
Por su parte, la Fundación Barcelona Macula aparece con el registro NCT05258032 en el registro de ensayos clínicos con un estudio de caracterización estructural y funcional de enfermedades oculares raras, incluida la de Best, y dirigido por el investigador Marc Biarnes Pérez y que estaría pendiente de presentar resultados.
Estudios en China y Oriente Medio
En China, la investigación sobre BEST1 se centra principalmente en estudios genético-clínicos observacionales de cohortes locales, sin ensayos terapéuticos activos. Similarmente, en Qatar y los países del Golfo, aunque hay creciente interés en terapias génicas para enfermedades retinianas hereditarias (estimuladas por alta prevalencia asociada a consanguinidad), no se han reportado ensayos específicos para enfermedad de Best. Tampoco se conocen avances en investigación en resto de Asia, Australia, África o América Latina ni Canadá. O al menos en registros accesibles.
Conclusión
El anuncio de Opus Genetics representa un punto de inflexión: la primera oportunidad clínica de intervención terapéutica específica para enfermedad de Best.
Mientras ese ensayo avanza en Estados Unidos, Europa consolida las bases científicas mediante estudios de historia natural y caracterización genética en cohortes grandes y bien documentadas.
En España, grupos clínicos avanzan en investigación fundamental en edición génica, acercando la posibilidad de terapias personalizadas.
Para los pacientes afectados, el horizonte, aunque aún lejano, es notablemente más esperanzador que hace tan solo dos años.
Para más información Para más información, visita www.opusgtx.com.
Artículo de elaboración propia, con ayuda de IA para análisis, y síntesis de documentos, así como para la redacción del artículo.




{kind=link}